
Experimental and modelling studies of binary mixtures:
how Molecular Dynamics simulations can help in understanding unexpected chemical-physical properties
时间:2024年7月10日(周三)下午15:40
地点:化学学科楼四楼报告厅
报告人简介:
Francesca Mocci is Aggr. Professor of Physical Methods in Organic Chemistry at the University of Cagliari (Italy) since 2007. She earned her Ph.D. in Chemistry from the University of Cagliari in 2002, working under the guidance of Prof. Giuseppe Saba. Over the years, she has served as a visiting professor at various institutions, including Stockholm University (2011–2016), Kosice Pavol Jozef Safarik University (2016), the Petru Poni Institute of Macromolecular Chemistry in Iaşi, Romania (2018–present), and Vilnius University (2023). She is in charge of the Computational Chemistry Laboratory at the Department of Chemical and Geological Sciences at Cagliari University and has taught molecular modelling in many international schools, supervised more than 20 Master and PhD students, both at Cagliari and Stockholm University, and hosted in her research group more than 30 students and researchers from across the world. She has been actively engaged in organizing various local, national, and international conferences and scientific schools, and served as the Chair of the International Conference of Theoretical Biophysics in 2015 (Cagliari). Currently, she holds the role of Training School Coordinator for the EU-funded Cost Action COSY.
She is Co-Editor of the volume “Molecular Dynamics Simulations and Reaction Rates” in the major reference work entitled “Comprehensive Computational Chemistry” of several volumes. She has served in the committee of Swedish Research Council for evaluation of research proposal in the field of physical chemistry. Her primary research focus revolves around computational investigations into conformational preferences, reactivity, and structural organization of organic and bio-organic systems, employing both classical and quantum mechanical modeling techniques. She has published over 60 papers in international peer-reviewed journals, and 10 book chapters.
报告摘要:
Liquid mixtures are widely used in many areas of chemistry, such as solvents in chemical synthesis or separation sciences. Typically, they are macroscopically fully miscible liquids, and the variation of many physical-chemical properties with the composition can be roughly predicted by knowing the properties of the components. However, this is not always the case. When unexpected behaviors are observed during the mixing of two solvents or when using binary mixtures to dissolve solutes, molecular modeling—particularly Molecular Dynamics simulations—constitutes a very powerful technique to understand the underlying reasons at the molecular level.
In this talk I will present few examples of combined experimental and computational studies of binary mixtures.
In the first, we use a combined experimental and modelling approach to study the structural organization of N-methyl-2-pyrrolidone + water mixtures. By a densitometry, x-ray diffraction, and molecular dynamics investigation, we explained why when adding water to an organic solvent having higher density than water density than water, the density increases instead of decreasing.

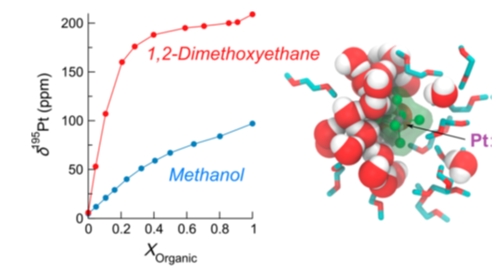
In the second one, I will present a combined NMR-MD approach aimed at understanding the solvation of the [PtCl6]2– anion in Water–Methanol and Water–Dimethoxyethane Binary Mixtures, and in particular why the 195Pt NMR varies with the organic component fraction in a very different way depending on the organic solvent used in the binary mixture.
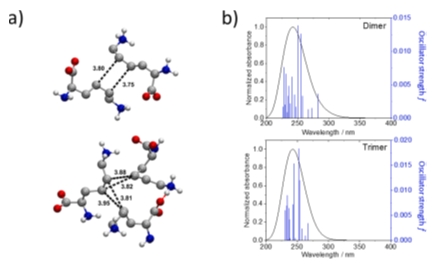
In the third one, I will focus on the investigation of the effect of aggregation on the optical property of L-lysine. While the zwitterionic nature of this amino acids at their natural isoelectric point could suggest that the aggregates, responsible for the optical properties are formed through ionic interactions and hydrogen bonds, TD-DFT calculations of UV absorption do not support this hypothesis. Due to the flexible nature of L-lysine, sampling the aggregates formed in aqueous solution at various concentrations is not a trivial task. In this last example, I will discuss how molecular dynamics simulation trajectories of model systems can be analysed to gain insight into the structure of the aggregates and describe some limitations of the method.


 当前位置:
当前位置:

